Other ligands
There are many different ligands encountered in organometallic chemistry. It is important to understand the electron donation/accepting properties of these ligands and which orbitals bond in which way.
These ligands aren’t always organometallics but can be explained by organometallic principles. Any lone pairs present are not stereochemically active. These lone pairs do not affect the arrangement of the other ligands only other ligands affect the stereochemistry of other ligands.
Nitrosyl complexes
Complexs of nitric oxides are not organometallic as there are no metal carbon bonds although they act very similarly to carbonyl ligands. These have a π* orbital that lies very close to the d orbitals on the metal which allowed back bonding to occur. The unpaired electron of free NO resides in a π* orbital and is easily removed to give NO+ which is isoelectronic to CO but a stronger π acceptor. This is a very strong back bonding agent which allows a strong bond to form.
NO contains one more electron and is bent when part of a one electron ligand, it can also be linear and becomes a three electron bonding mode. This is the same species that can form both 1 and 3 electron bonds. The addition of a ligand to a complex that already has 18 electrons can cause the 3 bonding mode to turn into a 1 bonding mode allowing the complex to remain stable.
The NO ligands donate three electrons each meaning there is a row of transition metals which are isoelectronic and isostructural to Ni(CO)4.
Cr(NO)4 Mn(NO)3(CO) Fe(NO)2(CO)2 Co(NO)(CO)3 Ni(CO)4.
Isocyanide complexes:
Nitrosyl complexes
Complexs of nitric oxides are not organometallic as there are no metal carbon bonds although they act very similarly to carbonyl ligands. These have a π* orbital that lies very close to the d orbitals on the metal which allowed back bonding to occur. The unpaired electron of free NO resides in a π* orbital and is easily removed to give NO+ which is isoelectronic to CO but a stronger π acceptor. This is a very strong back bonding agent which allows a strong bond to form.
NO contains one more electron and is bent when part of a one electron ligand, it can also be linear and becomes a three electron bonding mode. This is the same species that can form both 1 and 3 electron bonds. The addition of a ligand to a complex that already has 18 electrons can cause the 3 bonding mode to turn into a 1 bonding mode allowing the complex to remain stable.
The NO ligands donate three electrons each meaning there is a row of transition metals which are isoelectronic and isostructural to Ni(CO)4.
Cr(NO)4 Mn(NO)3(CO) Fe(NO)2(CO)2 Co(NO)(CO)3 Ni(CO)4.
Isocyanide complexes:
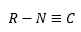
These complexes are isoethronic with CO although the carbon and the nitrogen are the other way round. These are stronger σ-donors than CO although it is a weaker π acceptor. It is a better σ donor due to the higher electron density as the nitrogen is less electronegative than oxygen. Isocyanide compounds are capable of stabilising higher oxidation states than CO. This relies heavily on donation and less on accepting meaning the metal centre has a higher electron density being donated.
Phosphine ligands
Phosphine ligands are two electron donors. These ligands are special as they give a lone pair of electrons to the metal centre the variation in the phosphine ligands means that a number of different factors can be controlled such as:
· Donor strength.
· Acceptor strength.
This can also be manipulated by the steric influence of the ligand by adding a varying range of substituents. This is based on the Tolman angle. The electronic properties can be systematically varied on a phosphine nickel tricarbonyl compound. This is purely based on the electronic properties of the phosphine so there are little steric effects caused as the nickel is a large atom and the carbonyl peaks are very small.
Phosphine ligands
Phosphine ligands are two electron donors. These ligands are special as they give a lone pair of electrons to the metal centre the variation in the phosphine ligands means that a number of different factors can be controlled such as:
· Donor strength.
· Acceptor strength.
This can also be manipulated by the steric influence of the ligand by adding a varying range of substituents. This is based on the Tolman angle. The electronic properties can be systematically varied on a phosphine nickel tricarbonyl compound. This is purely based on the electronic properties of the phosphine so there are little steric effects caused as the nickel is a large atom and the carbonyl peaks are very small.
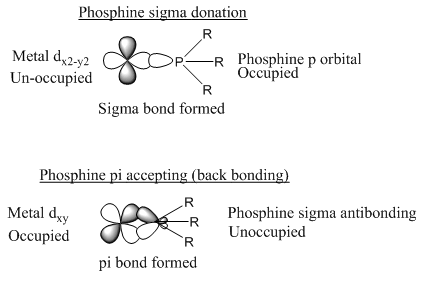
Phosphines are σ donors as well as being able to accept electron density into their P-C σ* orbitals. This has been thought to be vacant d orbitals but this is not true. These are σ* orbitals which have very similar shape to a p orbital. These have the right π symmetry to accept electrons from the metal centre. The phosphine is a much better donor and much worse acceptors than the carbonyl ligand. These properties can be varied by changing the R groups. By putting large bulky alkyl groups there will be a positive inductive effect. This would cause a large electron density into the phosphine group. This makes this a better donor but a worse acceptor.
The addition of electron withdrawing groups increases the π accepting character but reduces the σ donation character. The difference in stretching frequencies for the CO ligand measures the amount of donation of the phosphine ligand.
The steric properties of these ligands also give a measure of the alkyl ligand and how the umbrella of these alkyl groups form. The tolyl ligand is much bigger sterically as it has a steric group in its ortho position this creates a massive angle of 194°. There are not many phosphine ligands that can fit around the metal.
Alkene ligands
Alkene ligands can be reacted using a number of different addition. With the use of light or heat alkenes can be added to an 18 electron complex. These are not needed if the complex is under 18 electrons. Alkenes are two electron donors.
The addition of electron withdrawing groups increases the π accepting character but reduces the σ donation character. The difference in stretching frequencies for the CO ligand measures the amount of donation of the phosphine ligand.
The steric properties of these ligands also give a measure of the alkyl ligand and how the umbrella of these alkyl groups form. The tolyl ligand is much bigger sterically as it has a steric group in its ortho position this creates a massive angle of 194°. There are not many phosphine ligands that can fit around the metal.
Alkene ligands
Alkene ligands can be reacted using a number of different addition. With the use of light or heat alkenes can be added to an 18 electron complex. These are not needed if the complex is under 18 electrons. Alkenes are two electron donors.
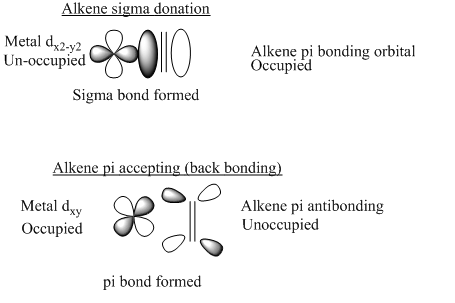
The antibonding interaction can be measured by measuring the bond strength of the alkene bond by using IR radiation or by measuring the bond length through x-ray crystallography. The alkene ligands are quite large as they sit very close to the metal centre. This means that it is possible for these to form complexes under 18 electrons.
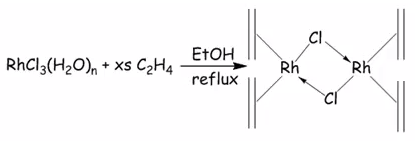
This is a 16 electron compounds and is rhodium (I) square planar compound. Rhodium has 9 electrons the alkenes have 2 electrons each and the dative and single bond create 16 electron atoms.
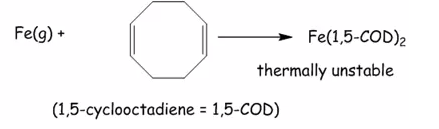
This is thermally unstable as it does have 18 electrons it only has 16 electrons.
The bonding of alkenes to transition metals occurs through both π donation and π accepting. The back donation weakens the carbon carbon bond causing a lengthening of the bond. The angles around the carbon are reduced from 120° to sp2 hybridised to sp3 hybridised carbon atom.
· increase in length
· increase in angle
Transition metal alkyl complexes
For a long time people thought that transition metal carbon σ bonds were intrinsically unstable. The electron count of platinum trimethyl iodide species is an eighteen electron species through triply bridging iodide ligands.
The thermodynamics of these species show some interesting strengths between the different carbon bond strengths. It can be seen that the transition metal complexes have an increasing bond strength to carbon down the group. This is due to better p-d orbital overlap as the energies are much more comparable. This is the opposite to main group trends as these orbitals become steadily more diffuse until there bonding no longer occurs.
The reason why transition metal groups are so unstable is due to the kinetic grounds of the reaction. There are a number of vacant orbitals that can be filled in the transition metal groups which allows reactions to occur quickly and easily. It is very easy for other ligands to quickly fill d orbitals causing a rapid antibonding interaction. The kinetic stability can be summed up in the β hydride formation. This is a reductive elimination of alkyl chains on the complex forming alkene groups.
The bonding of alkenes to transition metals occurs through both π donation and π accepting. The back donation weakens the carbon carbon bond causing a lengthening of the bond. The angles around the carbon are reduced from 120° to sp2 hybridised to sp3 hybridised carbon atom.
· increase in length
· increase in angle
Transition metal alkyl complexes
For a long time people thought that transition metal carbon σ bonds were intrinsically unstable. The electron count of platinum trimethyl iodide species is an eighteen electron species through triply bridging iodide ligands.
The thermodynamics of these species show some interesting strengths between the different carbon bond strengths. It can be seen that the transition metal complexes have an increasing bond strength to carbon down the group. This is due to better p-d orbital overlap as the energies are much more comparable. This is the opposite to main group trends as these orbitals become steadily more diffuse until there bonding no longer occurs.
The reason why transition metal groups are so unstable is due to the kinetic grounds of the reaction. There are a number of vacant orbitals that can be filled in the transition metal groups which allows reactions to occur quickly and easily. It is very easy for other ligands to quickly fill d orbitals causing a rapid antibonding interaction. The kinetic stability can be summed up in the β hydride formation. This is a reductive elimination of alkyl chains on the complex forming alkene groups.
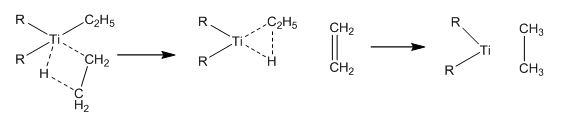
Stabilisation of transition metal complexes
This leaves the question on how these transition metal complexes are stabilised. One of the only methods this can occur is the blocking of coordination sites on the complex. This can be carried out by using large ligand groups that do not allow the bonding of any more ligands to the metal. Another way this can be carried out is only using ligands where β reductive elimination cannot occur. this means that the β carbon group cannot have any hydrogen atoms present.
Synthesis of transition metal alkyls can only occur through the insertion of the M-H bonds in oxidative addition. The metathesis reaction involves the alkylation of metal halides. This allows many different compounds to be formed so long as the alkyl attached does not have any β hydride atoms present.
Metal alkyls and the 18-electron rule, if a metal alkyl follows the 18 electron rule then it is classed as being stable. This is quite a simple process that is discussed thoroughly in other lecture notes. When complexes do not follow the 18 electron rule they need to be stabilised by other means. One of the best examples of this is the stabilisation of titanium complexes through the reaction of multiple bulky alkyl groups. This produces a steric hindrance which doesn’t allow the reactions of any other ligand groups.
The reactivity of the transition metal alkyl is usually through M-C bond cleavage this is very similar to the Grignard type reactions that have already been studied. It is important to remember that the carbon acts as a nucleophile meaning that the bond polarities need to be considered on each group for an accurate representation of how the reaction occurs.
Transition metal allyl complexes are formed through synthesis of allylic halides. This involve the reaction of these groups with the loss of the halide group forming a negative charge which then coordinates with the complex. This can either occur in a head on bonding formation or a side on bonding formation. These both allow the formation of a 3 or 1 coordination ligand.
The transition metal allyl complexes can be produced through protonation of dienes of metathesis with metal salts.
This leaves the question on how these transition metal complexes are stabilised. One of the only methods this can occur is the blocking of coordination sites on the complex. This can be carried out by using large ligand groups that do not allow the bonding of any more ligands to the metal. Another way this can be carried out is only using ligands where β reductive elimination cannot occur. this means that the β carbon group cannot have any hydrogen atoms present.
Synthesis of transition metal alkyls can only occur through the insertion of the M-H bonds in oxidative addition. The metathesis reaction involves the alkylation of metal halides. This allows many different compounds to be formed so long as the alkyl attached does not have any β hydride atoms present.
Metal alkyls and the 18-electron rule, if a metal alkyl follows the 18 electron rule then it is classed as being stable. This is quite a simple process that is discussed thoroughly in other lecture notes. When complexes do not follow the 18 electron rule they need to be stabilised by other means. One of the best examples of this is the stabilisation of titanium complexes through the reaction of multiple bulky alkyl groups. This produces a steric hindrance which doesn’t allow the reactions of any other ligand groups.
The reactivity of the transition metal alkyl is usually through M-C bond cleavage this is very similar to the Grignard type reactions that have already been studied. It is important to remember that the carbon acts as a nucleophile meaning that the bond polarities need to be considered on each group for an accurate representation of how the reaction occurs.
Transition metal allyl complexes are formed through synthesis of allylic halides. This involve the reaction of these groups with the loss of the halide group forming a negative charge which then coordinates with the complex. This can either occur in a head on bonding formation or a side on bonding formation. These both allow the formation of a 3 or 1 coordination ligand.
The transition metal allyl complexes can be produced through protonation of dienes of metathesis with metal salts.
