HPLC analysis is a highly popular technique. The chromatographic principles section should be referred to through this.
HPLC
High performance liquid chromatography was developed in the 1970s and has been used since. HPLC uses the interactions between molecules to cause separation. The two types of molecules that can be separated are either hydrophobic or hydrophilic interactions.
Hydrophobic interactions can be used to separate carbon chained molecules such as proteins or amino acids.
Hydrophilic interactions can be used to separate ionic materials, these can be acidified proteins or other partial ionised materials.
A HPLC system has multiple parts that are used to both prepare and analyse the sample. The initial system is the injection system. The injection system uses a needle to withdraw a small amount of the liquid sample to be analysed. It then takes this sample and injects it into the pump system.
The pump pushes the mobile phase and analyte into the column, this creates a large pressure (400 bar). This pressure is necessary to get the most separation amoungst the analytes present.
The analytes and the mobile phase then enters the column. The column is either hydrophobic or hydrophilic depending on the analysis needing to be carried out.
The molecules are usually analysed using UV spectroscopy, this is only effective if the analytes have a chromophore.
Mobile phase
An important feature of a HPLC is the vast array of mobile phases that can be used to aid a separation, a commonly used mobile phase is an organic solvent (ranging from 5-60%) with a buffer made up with water. In reversed phase chromatography the water can be seen as more a carrier, as the column will be an organic based column there will be no interactions between the water and the column allowing a straight flow through the column. The organic portion of the phase is important as this interacts with any hydrophobic materials on the stationary phase, drawing them through the column to elution. This is discussed in greater detail in the “column” section of these notes. Buffers added to the solution help to maintain pH, it is worth noting that these are salts, when changing the analysis of a column the column should be washed through with ca. 5 column volumes of mobile phase without the buffer. If there is a large change in solvent make up the buffer can precipitate out, ruining the column.
The solvent used must not interfere with the detection method used, this normally means the solvent used must not be too similar in any given properties with the analyte or the analysis is unlikely to be successful.
Modifiers are then added, these are usually trifluoroacetic acid (TFA) or N, N’-dimethylamine. The TFA acts as a non-coordinating acid. This helps to maintain the pH of a solution so there is not a rapid interchanging of any of the analyte properties through the analysis causing a different elution time.
The mobile phase to be used in the analysis needs to be tailored specifically to the analyte needing to be found. In HPLC there are two different mobile phases used. The mobile phases are mixed in different amounts, the different amounts of mobile phase cause different interactions with the analytes and the stationary phase.
The mobile phases are changed on a gradient scale, it starts with 100% mobile phase A. Mobile phase B is then added, this usually goes from 0-60% mobile phase B as this should wash all of the analytes present off of the column.
All solvents must be extremely pure. This is quite self-explanatory, any impurities will mess with analysis. There is also a large chance that impurities can cause blockages somewhere in the column. This means that with nearly all of the analysis there needs to be a filtering of the mobile phase before the HPLC column is used.
Degassing also occurs for the mobile phase as the large pressure differences during the analysis will cause any dissolved gas to form a bubble, creating a noisy spectrum.
Pump
The pump is a very important part of the system. A high pressure is needed to cause the largest amount of interactions between the mobile and the stationary phase. This puts a limit on the density of the stationary phase. With a high density stationary phase the pressure can build to a high limit causing damage to the pump. This is important to negate and can be negated by stronger pumps or a less dense stationary phase.
The pump is a hard material such as sapphire or emerald, which has little interaction with the mobile phase, the diameter of the pump is around 3 mm. This pumps into a well fitted seal, this is quite a fragile part of the machine, any grit will cause the seal to be lost and can lead to issues with the analysis.
Pumps give very precise control to the mobile phase flow rate. This is important as, over the course of many analytical runs, any inconstancies will ruin results. There can be two types of pump with either a single solvent used at a set rate in what is termed isocratic elution. This is the most simple of all HPLC methods in which a solvent is made up and constantly pumped through the column. Because of this an isocratic method is favourable but its simplicity makes it considerably less powerful, for any complex solutions the isocratic method doesn’t have the power to separate or remove some of the mobile phase components.
Two pumps are used if a gradient method is needed, this uses the ability for two mobile phases to be made of differing make ups, these can be pumped at the same time in different amounts giving a different overall mobile phase present in the pump. This allows a equilibriation time at the beginning of a run with a more hydrophilic phase to wash any unwanted ions off of the column, the mobile phase is slowly made more hydrophobic removing the analytes slowly from the column. This can then be ramped up to remove any strong organic binders and allowed to re-equilibrate to the beginning again.
Injection
Firstly the analyte needs to be injected into the coloum. This is easier said then done. It is almost impossible to inject into a system at around 400 bar. This means an injector that is only really seen on HPLC is used. This involves 6 ports in a circle. These ports are the: unjector, waste, column, solvent in (from pump), 2 sample loops.
The load position has the syringe filling the sample loops with sample to be analysed. The position shows that the mobile phase travels straight into the column. The sample loop does not run to waste as excactly the right amount to be injected is held in the sample loop. When the injector rotates the sample loops is then attached to the mobile phase and is back washed through into the column. This allows separation to occur and analysis to take place.
High performance liquid chromatography was developed in the 1970s and has been used since. HPLC uses the interactions between molecules to cause separation. The two types of molecules that can be separated are either hydrophobic or hydrophilic interactions.
Hydrophobic interactions can be used to separate carbon chained molecules such as proteins or amino acids.
Hydrophilic interactions can be used to separate ionic materials, these can be acidified proteins or other partial ionised materials.
A HPLC system has multiple parts that are used to both prepare and analyse the sample. The initial system is the injection system. The injection system uses a needle to withdraw a small amount of the liquid sample to be analysed. It then takes this sample and injects it into the pump system.
The pump pushes the mobile phase and analyte into the column, this creates a large pressure (400 bar). This pressure is necessary to get the most separation amoungst the analytes present.
The analytes and the mobile phase then enters the column. The column is either hydrophobic or hydrophilic depending on the analysis needing to be carried out.
The molecules are usually analysed using UV spectroscopy, this is only effective if the analytes have a chromophore.
Mobile phase
An important feature of a HPLC is the vast array of mobile phases that can be used to aid a separation, a commonly used mobile phase is an organic solvent (ranging from 5-60%) with a buffer made up with water. In reversed phase chromatography the water can be seen as more a carrier, as the column will be an organic based column there will be no interactions between the water and the column allowing a straight flow through the column. The organic portion of the phase is important as this interacts with any hydrophobic materials on the stationary phase, drawing them through the column to elution. This is discussed in greater detail in the “column” section of these notes. Buffers added to the solution help to maintain pH, it is worth noting that these are salts, when changing the analysis of a column the column should be washed through with ca. 5 column volumes of mobile phase without the buffer. If there is a large change in solvent make up the buffer can precipitate out, ruining the column.
The solvent used must not interfere with the detection method used, this normally means the solvent used must not be too similar in any given properties with the analyte or the analysis is unlikely to be successful.
Modifiers are then added, these are usually trifluoroacetic acid (TFA) or N, N’-dimethylamine. The TFA acts as a non-coordinating acid. This helps to maintain the pH of a solution so there is not a rapid interchanging of any of the analyte properties through the analysis causing a different elution time.
The mobile phase to be used in the analysis needs to be tailored specifically to the analyte needing to be found. In HPLC there are two different mobile phases used. The mobile phases are mixed in different amounts, the different amounts of mobile phase cause different interactions with the analytes and the stationary phase.
The mobile phases are changed on a gradient scale, it starts with 100% mobile phase A. Mobile phase B is then added, this usually goes from 0-60% mobile phase B as this should wash all of the analytes present off of the column.
All solvents must be extremely pure. This is quite self-explanatory, any impurities will mess with analysis. There is also a large chance that impurities can cause blockages somewhere in the column. This means that with nearly all of the analysis there needs to be a filtering of the mobile phase before the HPLC column is used.
Degassing also occurs for the mobile phase as the large pressure differences during the analysis will cause any dissolved gas to form a bubble, creating a noisy spectrum.
Pump
The pump is a very important part of the system. A high pressure is needed to cause the largest amount of interactions between the mobile and the stationary phase. This puts a limit on the density of the stationary phase. With a high density stationary phase the pressure can build to a high limit causing damage to the pump. This is important to negate and can be negated by stronger pumps or a less dense stationary phase.
The pump is a hard material such as sapphire or emerald, which has little interaction with the mobile phase, the diameter of the pump is around 3 mm. This pumps into a well fitted seal, this is quite a fragile part of the machine, any grit will cause the seal to be lost and can lead to issues with the analysis.
Pumps give very precise control to the mobile phase flow rate. This is important as, over the course of many analytical runs, any inconstancies will ruin results. There can be two types of pump with either a single solvent used at a set rate in what is termed isocratic elution. This is the most simple of all HPLC methods in which a solvent is made up and constantly pumped through the column. Because of this an isocratic method is favourable but its simplicity makes it considerably less powerful, for any complex solutions the isocratic method doesn’t have the power to separate or remove some of the mobile phase components.
Two pumps are used if a gradient method is needed, this uses the ability for two mobile phases to be made of differing make ups, these can be pumped at the same time in different amounts giving a different overall mobile phase present in the pump. This allows a equilibriation time at the beginning of a run with a more hydrophilic phase to wash any unwanted ions off of the column, the mobile phase is slowly made more hydrophobic removing the analytes slowly from the column. This can then be ramped up to remove any strong organic binders and allowed to re-equilibrate to the beginning again.
Injection
Firstly the analyte needs to be injected into the coloum. This is easier said then done. It is almost impossible to inject into a system at around 400 bar. This means an injector that is only really seen on HPLC is used. This involves 6 ports in a circle. These ports are the: unjector, waste, column, solvent in (from pump), 2 sample loops.
The load position has the syringe filling the sample loops with sample to be analysed. The position shows that the mobile phase travels straight into the column. The sample loop does not run to waste as excactly the right amount to be injected is held in the sample loop. When the injector rotates the sample loops is then attached to the mobile phase and is back washed through into the column. This allows separation to occur and analysis to take place.

Oven
An oven is sometimes used, as the retention time varies with temperature it can be important to maintain the column at a set temperature through the experiment. This normally means the column is equilibrated at slightly above ambient temperature.
Column
The column is the most important part of the HPLC device. The column can either be hydrophobic or hydrophilic and in varying amounts. The hydrophobic tendencies of the column can be increased or decreased depending on the length of the carbon chains attached to a silica membrane. It also depends on the size of the analyte. If the analyte is large, for example a protein, then a shorter molecule is used such as a C4 column rather than a C18 or C8 that will be used for an amino acid.
The column can be heated to allow different separation times. With an increase in temperature a better separation is found as well as a faster elution time. Although this can back fire if the analyte elutes close to the start of the run as this can cause analytes to be crushed together and therefore resolution to be lost.
The column can also be fitted with a guard column to partially protect the column although it can cause some of the separation and therefore resolution to be lost. It is safe to say the column is the most important part of the HPLC device and it is very important that the right column is chosen.
Column attachment
An oven is sometimes used, as the retention time varies with temperature it can be important to maintain the column at a set temperature through the experiment. This normally means the column is equilibrated at slightly above ambient temperature.
Column
The column is the most important part of the HPLC device. The column can either be hydrophobic or hydrophilic and in varying amounts. The hydrophobic tendencies of the column can be increased or decreased depending on the length of the carbon chains attached to a silica membrane. It also depends on the size of the analyte. If the analyte is large, for example a protein, then a shorter molecule is used such as a C4 column rather than a C18 or C8 that will be used for an amino acid.
The column can be heated to allow different separation times. With an increase in temperature a better separation is found as well as a faster elution time. Although this can back fire if the analyte elutes close to the start of the run as this can cause analytes to be crushed together and therefore resolution to be lost.
The column can also be fitted with a guard column to partially protect the column although it can cause some of the separation and therefore resolution to be lost. It is safe to say the column is the most important part of the HPLC device and it is very important that the right column is chosen.
Column attachment
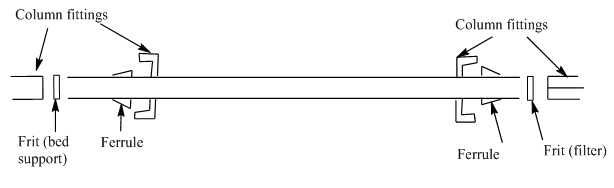
Column chemical makeup
The column is made up of either a hydrophobic or hydrophilic material. Most of the use in pharmaceuticals is centred on the reverse phase chromatography which uses a hydrophobic column. The columns produced all have the same base structure. The stationary phase is made of highly porous silica with a very high surface area of 100-500 m2g-1, so lots of adsoption sites are present for the analyte molecules. The silica groups are normally small spherical beads with a low size dispersity meaning they are all of similar size.
A silica column is usually used with silanol end groups. In normal phase chromatography these end groups are left in their usual silanol form. These are very hydrophilic, although there are issues due to the variation in interactions caused by the presence of hydrogen bonding in the system. This means that the pH of the run needs to be carefully monitored or the silanol groups will undergo a high dissociation. Some of the different silanol environments formed are shown below:
The column is made up of either a hydrophobic or hydrophilic material. Most of the use in pharmaceuticals is centred on the reverse phase chromatography which uses a hydrophobic column. The columns produced all have the same base structure. The stationary phase is made of highly porous silica with a very high surface area of 100-500 m2g-1, so lots of adsoption sites are present for the analyte molecules. The silica groups are normally small spherical beads with a low size dispersity meaning they are all of similar size.
A silica column is usually used with silanol end groups. In normal phase chromatography these end groups are left in their usual silanol form. These are very hydrophilic, although there are issues due to the variation in interactions caused by the presence of hydrogen bonding in the system. This means that the pH of the run needs to be carefully monitored or the silanol groups will undergo a high dissociation. Some of the different silanol environments formed are shown below:
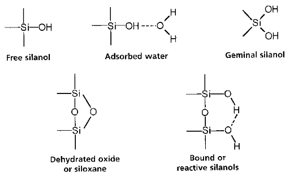
This shows a great variation in the electron environments that can be present in the silanol groups. For reversed phase chromatography a hydrophobic molecule is reacted with the silanol group creating an alkyl stationary phase.
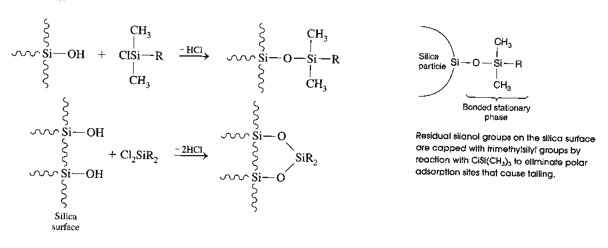
This shows how the polarity of the column has been completely reversed. Hence “reversed phase chromatography”.
There is a great importance of what is then bonded to this silica group. The table below shows the difference in the polarity of the columns and the polarity of the solvents:
There is a great importance of what is then bonded to this silica group. The table below shows the difference in the polarity of the columns and the polarity of the solvents:
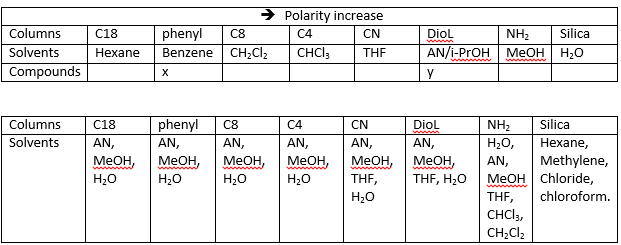
This can be thought as that the large carbon chains give a very large hydrophobic area. This means that in reversed phase chromatography a C18 column can be used to effectively separate the less polar compounds especially when small, such as amino acids.
The solvents that are used in the analysis need to be carefully considered. To think of a good example assume a gradient run is being carried out with mobile phase A being more hydrophilic than mobile phase B on a C18 column. The analyte in the example is a moderately hydrophobic oligonucleotide.
Mobile phase A tends to be a highly polar solution consisting of water mixed with a small amount 1-5% acetonitrile solution. This quickly washes any un-wanted polar molecules off of the column. The addition of a small amount of acetonitrile is to still have some hydrophobic species in the column to interact with the stationary phase, too much water can cause phase collapse due to the hydrophobic stationary phase. Over the gradient run the concentration of mobile phase B, consisting of water mixed with acetonitrile (60%) will be added. This causes a slow increase in acetonitrile concentration, this means that the oligonucleotide will begin to move down the column as it is being displaced on the stationary phase by acetonitrile molecules. This causes the analyte to elute. Once the oligoneclotide has eluted then the mobile phase may be rapidly increased to the 60% acetonitrile mark to wash any more hydrophobic materials off of the column. Once this has been done the mobile phase is reverted back to 5% acetonitrile and left to equilibrate.
Most of the discussion has been about reversed phase chromatography. This is for good reason as the reversed columns tend to be more stable as they do not have the issues related to the free silanol groups. The column is also much less sensitive to changes in the mobile phase. This means that small changes in the mobile phase can cause a large difference making the use of a gradient much harder as the mixing needs to be much more accurate leading to a larger error over time. With a reversed phase system the gradient will move from ~20-80% through the analysis. Compare this to the gradient for normal phase being ~1-5%.
It is known that like attracts like when it comes to polarity and charge. This shows that there needs to be attraction between the two components that need to be separated. This means that the mobile phase needs to be made similar to one of the components and the stationary phase close to the other component.
Components
The components being measured can also be analysed to see how their interactions will vary depending on the polarity of the stationary and mobile phases. Many predictions can be made using computer programs in a dry-lab format to allow a good prediction of the elution times. This allows a large amount of optimisation.
Particle size
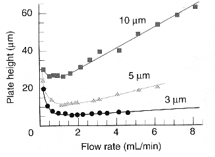
The particle size of the stationary phase is important to the different groups that are available for the different particle size. This allows different separation methods to be used allowing more effective or quicker separation. The theoretical plate height varies greatly depending on the amount size of the column and the ability to maintain the higher flow rate although maintaining the separation. This allows the most even interactions between the different surfaces that are present. The typical size of these beads are between 1.7 μm to 10 μm for most analytical methods.
The basis of the UHPC machines is to allow a faster separation to be carried out as the particle size is much smaller, which can be allowed due to an increase in pressure.
The solvents that are used in the analysis need to be carefully considered. To think of a good example assume a gradient run is being carried out with mobile phase A being more hydrophilic than mobile phase B on a C18 column. The analyte in the example is a moderately hydrophobic oligonucleotide.
Mobile phase A tends to be a highly polar solution consisting of water mixed with a small amount 1-5% acetonitrile solution. This quickly washes any un-wanted polar molecules off of the column. The addition of a small amount of acetonitrile is to still have some hydrophobic species in the column to interact with the stationary phase, too much water can cause phase collapse due to the hydrophobic stationary phase. Over the gradient run the concentration of mobile phase B, consisting of water mixed with acetonitrile (60%) will be added. This causes a slow increase in acetonitrile concentration, this means that the oligonucleotide will begin to move down the column as it is being displaced on the stationary phase by acetonitrile molecules. This causes the analyte to elute. Once the oligoneclotide has eluted then the mobile phase may be rapidly increased to the 60% acetonitrile mark to wash any more hydrophobic materials off of the column. Once this has been done the mobile phase is reverted back to 5% acetonitrile and left to equilibrate.
Most of the discussion has been about reversed phase chromatography. This is for good reason as the reversed columns tend to be more stable as they do not have the issues related to the free silanol groups. The column is also much less sensitive to changes in the mobile phase. This means that small changes in the mobile phase can cause a large difference making the use of a gradient much harder as the mixing needs to be much more accurate leading to a larger error over time. With a reversed phase system the gradient will move from ~20-80% through the analysis. Compare this to the gradient for normal phase being ~1-5%.
It is known that like attracts like when it comes to polarity and charge. This shows that there needs to be attraction between the two components that need to be separated. This means that the mobile phase needs to be made similar to one of the components and the stationary phase close to the other component.
Components
The components being measured can also be analysed to see how their interactions will vary depending on the polarity of the stationary and mobile phases. Many predictions can be made using computer programs in a dry-lab format to allow a good prediction of the elution times. This allows a large amount of optimisation.
Particle size
The particle size of the stationary phase is important to the different groups that are available for the different particle size. This allows different separation methods to be used allowing more effective or quicker separation. The theoretical plate height varies greatly depending on the amount size of the column and the ability to maintain the higher flow rate although maintaining the separation. This allows the most even interactions between the different surfaces that are present. The typical size of these beads are between 1.7 μm to 10 μm for most analytical methods.
The basis of the UHPC machines is to allow a faster separation to be carried out as the particle size is much smaller, which can be allowed due to an increase in pressure.
Column fragility and health
HPLC columns are usually seen as being fragile pieces of equipment that regularly undergo issues. This is an underserved reputation and columns can have a long life span depending on how well they are treated and what analysis that they are going.
There are a number of different aspects that can damage column efficiency. It is important to reference efficiency as there can be a slow decrease of efficiency on one form of analysis but the column can be easily transferred to the next analysis. This can lead to the ability increase column life by a large amount.
The five main issues that usually occur are: loss (gradual or instant) of bonding phase. Dissolving of the column surface. Binding materials to the column. Pressure increase. Column channelling.
Loss of bonding phase.
The loss of the bonding phase can occur for a number of different reasons the main reasons that are usually seen are that the pH of the solution being analysed falls below 2.0. When this occurs the silanol end groups tend to lose their hydrogen atoms becoming increasingly ionic in character. If this occurs there is less chance of the separation being carried out effectively. It is recommended to use buffers in this reaction process. High temperature can produce a number of different effects which include the increased solubility of the silica packing. This means that the number of different temperatures that are available to act as a variable are very limited before the column can undergo damage. As the silica packing dissolves there is an increase in peak width especially in the tailing peaks.
Dissolving Packing Material- End Voids.
At high pH normally above 8.0 can dissolve a large section of the silica phase which can rapidly form and end void. With a formation of end voids there can be a broadening or formation of rabbit eared peaks. There is the possibility of re filling these end voids with more packing materials. Salt solutions with concentrations above 200 mM tend to erode column beds by increasing the ionization and therefore the solubility of the silica present. There have been methods in which cutting the end void areas down slowly can occur.
Bound material
Material can bond to the packing over time forming different properties for the column and therefore some distinct changes in column make up. Uncharged nonpolar organics can stick to the column over time. These can cause the organic materials to broaden in peak size and then disappear completely. This can be due to water contamination, as small non-polar materials can begin to build up along the pores. When there is a build-up of bound material it is important to recognise that there needs to be a column wash. This can be carried out using highly concentrated acetonitrile although it is important to note that if a buffer is being used then the buffer needs to be removed before any large washing occurs otherwise the production of salts could occur. This can also be carried out by washing all the way to hexane and back. This will allow all of the organics to be removed although it is time consuming.
Another factor can be the slow celation of metal ions these can be removed with another chelating ion such as oxalate. If there are ion pair reagents the bonding and chelating effects usually cause the column to be of little use apart from ion pair chromatogram runs. The columns can be recovered with a large amount of washings.
Pressure increases
The next issue is an overly high pressure present the first step is to locate the pressure increase. This is discussed in most trouble shooting sections although here the column pressure will be discussed here. The increase in column pressure can be due to the inlet frit, the outlet frit and the column bed. The most likely problem is the inlet frit as this has a very small particle filter size. Replacing the frit is the last result as there can be a large amount of damage to the column in the form of lost packing material. This means it is better to try other unblocking methods such as column reversal. In many cases a blocked column is either a very tricky fix or the death of the column. The frit can either be replaced or washed in a sonicator with nitric acid (6N).
Column channelling
The column channelling is also known as “centre voiding” this is where there is a void in the centre of the column which cannot be replaced. The only known method is hitting the column on a desk twice and either end, reversing it at a high flow rate and then carrying on. This has been known to work in many cases and is a good last resort test.
Detection
The detection of the molecules as they elute from the column they are analysed usually by UV spectroscopy. This means the components coming off of the column need to have a chromophore attached to them. Many analytes do not have a chromophore so have one added in a derivatization reaction. This can be un-reliable and for large component mixtures the derivatization may or may not work.
UV-Vis
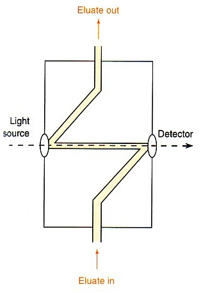
All of the processes involved in UV-Vis analysis are the same in ordinary UV-Vis analysis. The UV-Vis spectroscope is placed at one end of a short flow tube, normally about 5μL volume. This has a set path length of 1-5 mm and measures the absorption of the sample.
HPLC columns are usually seen as being fragile pieces of equipment that regularly undergo issues. This is an underserved reputation and columns can have a long life span depending on how well they are treated and what analysis that they are going.
There are a number of different aspects that can damage column efficiency. It is important to reference efficiency as there can be a slow decrease of efficiency on one form of analysis but the column can be easily transferred to the next analysis. This can lead to the ability increase column life by a large amount.
The five main issues that usually occur are: loss (gradual or instant) of bonding phase. Dissolving of the column surface. Binding materials to the column. Pressure increase. Column channelling.
Loss of bonding phase.
The loss of the bonding phase can occur for a number of different reasons the main reasons that are usually seen are that the pH of the solution being analysed falls below 2.0. When this occurs the silanol end groups tend to lose their hydrogen atoms becoming increasingly ionic in character. If this occurs there is less chance of the separation being carried out effectively. It is recommended to use buffers in this reaction process. High temperature can produce a number of different effects which include the increased solubility of the silica packing. This means that the number of different temperatures that are available to act as a variable are very limited before the column can undergo damage. As the silica packing dissolves there is an increase in peak width especially in the tailing peaks.
Dissolving Packing Material- End Voids.
At high pH normally above 8.0 can dissolve a large section of the silica phase which can rapidly form and end void. With a formation of end voids there can be a broadening or formation of rabbit eared peaks. There is the possibility of re filling these end voids with more packing materials. Salt solutions with concentrations above 200 mM tend to erode column beds by increasing the ionization and therefore the solubility of the silica present. There have been methods in which cutting the end void areas down slowly can occur.
Bound material
Material can bond to the packing over time forming different properties for the column and therefore some distinct changes in column make up. Uncharged nonpolar organics can stick to the column over time. These can cause the organic materials to broaden in peak size and then disappear completely. This can be due to water contamination, as small non-polar materials can begin to build up along the pores. When there is a build-up of bound material it is important to recognise that there needs to be a column wash. This can be carried out using highly concentrated acetonitrile although it is important to note that if a buffer is being used then the buffer needs to be removed before any large washing occurs otherwise the production of salts could occur. This can also be carried out by washing all the way to hexane and back. This will allow all of the organics to be removed although it is time consuming.
Another factor can be the slow celation of metal ions these can be removed with another chelating ion such as oxalate. If there are ion pair reagents the bonding and chelating effects usually cause the column to be of little use apart from ion pair chromatogram runs. The columns can be recovered with a large amount of washings.
Pressure increases
The next issue is an overly high pressure present the first step is to locate the pressure increase. This is discussed in most trouble shooting sections although here the column pressure will be discussed here. The increase in column pressure can be due to the inlet frit, the outlet frit and the column bed. The most likely problem is the inlet frit as this has a very small particle filter size. Replacing the frit is the last result as there can be a large amount of damage to the column in the form of lost packing material. This means it is better to try other unblocking methods such as column reversal. In many cases a blocked column is either a very tricky fix or the death of the column. The frit can either be replaced or washed in a sonicator with nitric acid (6N).
Column channelling
The column channelling is also known as “centre voiding” this is where there is a void in the centre of the column which cannot be replaced. The only known method is hitting the column on a desk twice and either end, reversing it at a high flow rate and then carrying on. This has been known to work in many cases and is a good last resort test.
Detection
The detection of the molecules as they elute from the column they are analysed usually by UV spectroscopy. This means the components coming off of the column need to have a chromophore attached to them. Many analytes do not have a chromophore so have one added in a derivatization reaction. This can be un-reliable and for large component mixtures the derivatization may or may not work.
UV-Vis
All of the processes involved in UV-Vis analysis are the same in ordinary UV-Vis analysis. The UV-Vis spectroscope is placed at one end of a short flow tube, normally about 5μL volume. This has a set path length of 1-5 mm and measures the absorption of the sample.
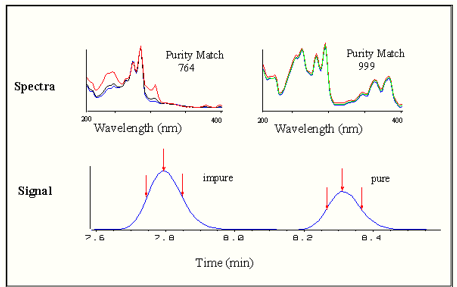
Comparison of a spectrum at three different elution times across all wavelengths can show any inconsistencies with the analyte and the pure material. If the spectrums taken at each retention time are identical then the sample is deemed to be pure.
Refractive index detectors
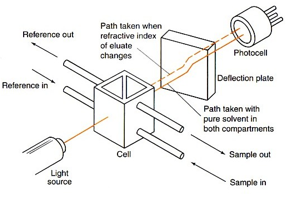
These detectors work by measuring the universal property of refractive index. This varies depending on the composition of the sample which means any change in composition can be measured. The only issue with this is the fact that the method is 1000 times less sensitive than UV. This causes the solvent to be very near saturation for a correct measurement to be made. It is also sensitive to any gradient, temperature and pressure changes. This means it is no good for most runs that need to be carried out.
Polymers are normally analysed by refractive index detectors.
These detectors work by measuring the universal property of refractive index. This varies depending on the composition of the sample which means any change in composition can be measured. The only issue with this is the fact that the method is 1000 times less sensitive than UV. This causes the solvent to be very near saturation for a correct measurement to be made. It is also sensitive to any gradient, temperature and pressure changes. This means it is no good for most runs that need to be carried out.
Polymers are normally analysed by refractive index detectors.
It is common for a mass spectrometer to be added to a HPLC machine. This usually needs a resolution of around 1.5 but is most effective at 2.0.
Sample preparation
Sample must be soluble in the mobile phase and have good enough interactions with the stationary phase for some retention to be present. If the sample is overloaded, at too high a concentration, the column can sustain damage.
The concentration of key analytes should be linear range of detector and the amount of sample introduced to the system depends on the size of the sample loop. Separated compounds can be collected and isolated after detection point.
Method development
The column chosen must be of the right material for the best separation, correct length and particle size are important.
Temperature
The temperature can be changed causing large differences in the chromatographic methods. The changes are caused by a variety of effects some of which can be easily predicted with the current components some of which must be determined empirically.
As the temperature increases there is generally an effect on the equilibrium constants of both the analyte and the mobile phase with the stationary phase. This can be easily predicted in some instances depending on the analyte and solvent being used. The dielectric constant will also change depending on the temperature causing a significant change in the polarity on occasion. This should be watched out for as elution times of certain species can have a large effect. The conformation changes of any components and any changes in the degree of solvation should also be considered. Thankfully a number of these systems have fully developed drylab predictions.
With an increase in temperature there is also a decrease in viscosity and faster column interactions. This means a better separation is achieved meaning an increase in plate height for each separation.
Mobile phase must be the right make up and the pH must be controlled.
Sample preparation
Sample must be soluble in the mobile phase and have good enough interactions with the stationary phase for some retention to be present. If the sample is overloaded, at too high a concentration, the column can sustain damage.
The concentration of key analytes should be linear range of detector and the amount of sample introduced to the system depends on the size of the sample loop. Separated compounds can be collected and isolated after detection point.
Method development
The column chosen must be of the right material for the best separation, correct length and particle size are important.
Temperature
The temperature can be changed causing large differences in the chromatographic methods. The changes are caused by a variety of effects some of which can be easily predicted with the current components some of which must be determined empirically.
As the temperature increases there is generally an effect on the equilibrium constants of both the analyte and the mobile phase with the stationary phase. This can be easily predicted in some instances depending on the analyte and solvent being used. The dielectric constant will also change depending on the temperature causing a significant change in the polarity on occasion. This should be watched out for as elution times of certain species can have a large effect. The conformation changes of any components and any changes in the degree of solvation should also be considered. Thankfully a number of these systems have fully developed drylab predictions.
With an increase in temperature there is also a decrease in viscosity and faster column interactions. This means a better separation is achieved meaning an increase in plate height for each separation.
Mobile phase must be the right make up and the pH must be controlled.
